Источник:
Международный неврологический журнал,
№3 2011г.
Донецкий областной детский клинический центр нейрореабилитации
Роль КАРНИЭЛЯ
в комплексной терапии
прогрессирующих мышечных дистрофий
у детей
С.К. Евтушенко, М.Р. Шаймурзин, И.С. Евтушенко
С целью патогенетического лечения вторичных метаболических нарушений при прогрессирующих мышечных дистрофиях у детей разработана методика курсовой терапии энергостимулирующего препарата Карниэль. Обследовано 40 детей с различными формами прогрессирующих мышечных дистрофий, из них 20 детей принимали Карниэль в составе комплексной терапии, 20 детей принимали стандартную схему лечения, но без Карниэля. Клинический эффект оказался достоверно выше в группе пациентов, принимавших Карниэль, чем в группе детей принимавших стандартную схему без Карниэля. Полученные данные следует учитывать в назначении долговременной курсовой терапии прогрессирующих мышечных дистрофий.
Нейро-мышечные заболевания (НМЗ) – большая гетерогенная в
клинико-генетическом аспекте группа прогрессирующих, практически
всегда приводящих к инвалидизации, наследственно-дегенеративных
заболеваний нервной системы, в основе которых лежит генетически
детерминированное поражение определенной структуры двигательной
единицы (мотонейрон спинного мозга, периферический нерв, мышечное
волокно).
В настоящее время большая часть НМЗ трудно курабельные и
неизличимы. Вместе с тем, данные клинического осмотра, комплексное
проведение ЭМНГ, включая ЭМГ-мониторинг, ЭКГ, ЭХО-КГ, заключение
генетика позволяет не только заподозрить, но и вовремя на ранних
этапах определить топическую лока лизацию патологического
процесса (мотонейрон спинного мозга, периферический нерв, мышечное
волокно), преимущественное распределение атрофии, слабости,
характер течения патологического процесса (медленно или быстро
прогрессирующее), диагностировать на ранних стадиях болезни
поражение сердечно-сосудистой, костно-суставной системы,
определить стадию кардиомиопатии, что дает возможность
спрогнозировать дальнейшее развитие заболевания, подобрать
патогенетически и симтоматически обоснованную терапию и попытаться
максимально затормозить прогрессирование патологического
процесса.
В связи с неуклонным увеличением числа детей с данной патологией
в 1997 году на базе Областного детского клинического центра
нейрореабилитации выделены амбулаторный прием и самостоятельные 4
специализированные стационарные койки для детей с нейро-мышечной
патологией. С 2001 года открыт современный ЭМГ-кабинет,
модифицирована клиническая лаборатория, с 2005 года открыт кабинет
ультразвуковой диагностики с проведением Эхо-КГ.
Для диагностики ранних проявлений прогрессирующих мышечных
дистрофий и амиотрофий, их фенокопий сотрудниками Областного
клинического детского центра нейрореабилитации модифицирован
диагностический и лечебный паттерн, адаптированный к практическому
здравоохранению, который включает в себя:
-
Клинический осмотр с использованием модифицированной шкалы
эффективности реабилитации детей с нейро-мышечной патологией;
-
Биохимическое исследование ферментов крови (АЛТ, АСТ, КФК, ЛДГ)
на аппарате «Фотометр-КФК-3»; исследование иммунологического
статуса (Ts, Th, T, CD25, CD4/CD8, IgG, Ig A, Ig E).
-
Электронейромиография с помощью компьютерного электромиографа
«Нейро-МВП-микро» фирмы «Нейрософт» (Россия)
-
Электрокардиография (аппарат «ЭК1К-01») при необходимости
холтеровский мониторинг ЭКГ с измерением АД.
- ЭХО-КГ ( аппарат «Logic 200 ProSeries»).
-
Исследование функции внешнего дыхания с помощью аппарата
Спиросефт-3000 (Япония).
-
Консультация ортопеда, педиатра, кардиолога, психиатра (по
показаниям);
-
Биопсия мышц в затруднительных случаях дифференциальной
диагностики первичных и вторичных мышечных дистрофий.
-
При затруднении дифдиагностики проводится МРТ головного,
спинного мозга (аппарат «Gyrocsan Intera T10» Голландия) –
ДОКТМО
-
Для выявления сочетанных сосудистых аномалий проводится УЗДГ
(аппарат «Logidop-4» фирмы «Kranzbuhler»), цветное дуплексное
сканирование (аппарат «Sanoline Elegre advanced», Simens).
-
Для исключения остеопороза, что достаточно часто выявляетя у
детей с НМЗ, проводится денсинометрия костей (рентгеновский
денсинометр двуэнергетический фирмы «General electric») – Донецкий
институт травматологи.
По частоте встречаемости прогрессирующие мышечные дистрофии
занимают ведущее место среди всех нейро-мышечных заболеваниях.
Прогрессирующие мышечные дистрофии (ПМД) – многочисленная группа
моногенных наследственных болезней нервной системы,
характеризующееся прогрессирующим снижением массы скелетных мышц,
ослаблением мышечной функции, снижением силы, сократительной
способности, обусловленное первичным дефектом в мышечных волокнах
и как следствие – развитие вторичных дисмета болических,
митохондриальных нарушений, снижение защиты от окислительного
стресса и пр. в конечном приводящее к жировой инфильтрации и
замещению соединительной тканью.
-
Основываясь на мировые, отечественные достижения в области
нейромиологии, а также собственных разработок в Центре внедрены,
адаптированы и предложены для практического врача стандарты лечения
детей с ПМД [2].
-
Сбалансированное лечебное питание (продукты, содержащие белок,
полиненасыщенные жирные кислоты, витамины, микроэлементы): овощи,
творог, морская рыба, печень. Весной и осенью курсовой прием
поливитаминных препаратов и микроэлементов: нейровитан, кидди
фарматон, мильгамма.
-
Щадящий массаж мышц с применением трофических мазей
(«Хондроксид», актовегиновая мазь, бальзам «Живокост»).
-
Дозированная лечебная физкультура с элементами
strench-гимнастики, направленная на поддержания и максимальное
сохранение функциональной способности не вовлеченных в
патологический процесс мышц в каждом конкретном случае с учетом
формы нейро-мышечного заболевания.
-
В комплекс лечения пневмопатий включены: синглетно-кислородная
терапия, дыхательная гимнастика, массаж дыхательной
мускулатуры.
-
Сопутствующая патология со стороны опорно-двигательного
аппарата: деформация туловища, позвоночника (нейро-мышечный
сколиоз) – увеличивает тяжесть дыхательных расстройств, в связи с
чем в Центре разработана методика мепоказана физиотерапевтические
методы, имитирующие элементы легкой маханического легкого массажа
на специальной механизированной кровати без подогрева («Relax»,
Япония). После чего проводится вакуумная стимуляция мышечного
корсета спины на аппарате «Вакотрон» («Vac BTL», Киев, Украина)
детям от 1 мес. До 3 лет. Детям от 3 лет и старше проводится
щадящая электромиостимуляция сохранных мышц на аппарате «АЭСТ-01-8»
(режимы: общемодулирующий и тренировка выносливости, длительность –
от 5 до 8 мин).
-
С целью укрепления мышечного корсета спины применяетя
сегментарные паравертебральная фармакопунктура с введением микродоз
препарата (нейромидин, актовегин, цераксон).
-
Для профилактики контрактур применяются специальные шины,
валики, фиксация конечностей в физиологическом положении на ночь с
использованием туторов, с целью адаптации передвижения с
оптимальной коррекцией деформаций используются стельки,
ортопедическая обувь, надколенники. Показано 1-2 часовое ношение
реклинаторов, корсетов в моменты наибольшей нагрузки на позвоночный
столб (сидение, ходьба и др.). В стадии декомпенсации ношение
корсетов практически постоянно, поскольку клинически имеет место
выраженная атония и гипотрофия мышечного корсета, влекущая за собой
резкую деформацию позвоночного столба, приводящую к вторичной
висцеропатии, ухудшению работы сердца, лёгких, влекущее за собой
ещё большую декомпенсацию патологического процесса.
-
При доброкачественных формах прогрессирующих мышечных дистрофий
(ПМД Бетлема, Ульриха, ранние стадии Эмери-Дрейфуса, Беккера) в
стадии стойкой компенсации, совместно с неврологами и ортопедами
решается вопрос об оперативном вмешательстве: тендо-,
миопластика.
-
Наличие остеопороза, подтвержденное денсинометрией обуславливает
назначение кальцийсодержищих препаратов: кальций D3 (кальцемин
адванс) длительностью до 6 мес. затем 1 мес. перерыв, в этот
промежуток диета, обогащенная солями кальция (нежирный творог,
сыворотка, твердые сорта сыра) под обязательным контролем
ионизированного кальция крови и УЗИ почек.
-
В связи с частыми ОРВИ, особенно в зимний период необходимо
назначение препаратов, влияющих непосредственно на
иммунокомпентные клетки и центральные механизмы регуляции
иммунитета, через которые оказывается вторичное
иммуностимулирующее влияние на организм, назначается:
бронхомунал-П (до 12 лет – по 3,5 мг/сут, с 12 лет -по 7,0 мг/сут) 10 дней прием, 20 дней перерыв. Курсы №.3.
-
При дилатационных кардиомиопатиях проводится курсовое
внутривенное введение кардиотрофических препаратов: неотон 1-2 г
или 10% раствор карнитина хлорид 0,5-1 г [3].
И все же, у детей с прогрессирущими мышечными дистрофиями биохимические изменения напрямую сопряжены со
вторичными нарушениями в цикле Кребса, выражающеея в гипофункции
митохондриальных ферментов, снижением содержания митохондриального
белка и общего объема митохондрий. В частности, по мере
прогрессирования миодистрофического процесса постепенно снижается
гликогенолиз (фосфорилазы) и гликолиз (гексокиназа и
лактатдегидрогеназа). Для компенсации гликогенолиза вовлеченная в
патологический процесс мышца усиливает процессы глюконеогенеза, при
этом используя энергию деградации жирных кислот в митохондриях, –
поскольку активность
3-гидроксиацил-КоА-дегидрогеназы остается достаточно высокой.
Вместе с тем, значительное увеличение содержания в пораженных
мышцах переносчиков глюкозы, ускоряет транспорт глюкозы в клетку,
что также усиливает процессы глюконеогенеза [4], в результате чего
снижается доступность окисленных коферментов, что приводит к
значительному снижению скорости окисления и накоплению
промежуточных продуктов обмена. При этом превалирующая часть
свободного карнитина превращается в ацилкарнитины и не участвует в
образова нии ацетил-СоА, участвующего в цикле трикарбоновых
кислот. Все вышеописанные биохимические изменения в миоцитах при
миодистрофии усиливают апоптоз в скелетных мышцах, усугубляющее
патологический процесс при миодистрофии.
Cущественную роль в патофизиологии
миодистрофического процесса играет ускорение апоптоза скелетных
мышц, вызванное хроническим накоплением окислительного стресса и
повреждений, митохондриальной дисфункцией. С другой стороны, такие
естественные воздействия, как синглетно-кислородная терапия,
направленной в альтернативной доставке кислорода в клетки и ткани
организма, дыхательная гимнастика, низкая калорийность пищи и
регулярные дозированные физические упражнения, препараты, механизм
действия которых сопряжен с циклом Креббса могут повлиять на
сигнальные пути апоптоза, ослабляя апоптозный сигналинг,
включающего митохондриальный и цитокинорецепторный, а также
стресс-индуцируемый пути апоптоза [5].
Поэтому необходимо курсовое и долговременное применение
препаратов, направленных на энергетическое обеспечение сохранной
мышечной ткани, функциональная активность которых сопряжена с
энергетическим пулом ацетил-КоА в митохондриях. Препараты
«метаболического действия» назначаются в сочетании или отдельно,
курсами до 3 месяцев 2 раз в год.
На основании нашего 10-летнего опыта лечения НМЗ мы попытались
подобрать лекарственное вещество, способный максимально
оптимимизировать нарушенные энерго-метаболические функции клетки.
Международные эксперты предлагают применять препараты, содержащие
левокарнитин. В течение ряда мы применяли элькар, в настоящее на
рынке появился его аналог карниэль, препарат зарегитрирован в
Украине.
Действующее вещество 20% раствора Карниэля /Элькара/ – левокарнитин.
L-карнитин (от лат. "carnis" – мясо.) выделен русскими учеными
Гулевичем и Кримсбергом в 1905 году из мясного экстракта.
L-карнитин в большом количестве содержится в красном мясе, рыбе,
домашней птице, молочных продуктах, крабовом мясе, авокадо [1].
20% раствор Карниэля /Элькара/ содержит природное вещество, родственное
витаминам группы В. Является кофактором метаболических процессов,
обеспечивающих поддержание активности КоА. Способствует
проникновению через мембраны митохондрий и расщеплению
длинноцепочных жирных кислот (пальмитиновой и др.) с образованием
ацетил-КоА (необходим для обеспечения активности
пируваткарбоксилазы в процессе глюконеогенеза, образования
кетоновых тел, синтеза холина и его эфиров, окислительного
фосфорилирования и образования АТФ). Мобилизует жир (наличие 3
лабильных метильных групп) из жировых депо. Конкурентно вытесняя
глюкозу, включает жирнокислотный метаболический шунт, активность
которого не лимитирована кислородом (в отличие от аэробного
гликолиза). Карнитин способствует удалению короткоцепочечных жирных
кислот из митохондрии, освобождая внутримитохондриальной СоА,
стабилизация уровня которого является важным для поддержания
клеточного энергетического метаболизма. Одной из важных функций
карнитина является контроль и модуляция внутриклеточного пула CоASH
[1].
CоASH является главнейшим метаболитом клетки: участвует в
биохимических процессах образования и деградации жирных кислот,
стероидов, фосфолипидов, синтезе ацетилхолина, окисления
пировиноградной кислоты, принимает участие в процессах катаболизма
органических кислот, ксенобиотиков [6].
В ряде исследований выявлены защитные функции при апоптозе.
Протективные свойства L-карнитина обусловлены ингибированием
церамидов, являющихся мощными промторами клеточного апоптоза, и
каспаз, являюшихся ключевыми медиаторами апоптоза [5].
Карниэль /Элькар/ 20% в 1 мл содержит 200 мг левокарнитина и принимался
перорально за 30 минут до еды (дополнительно разбавлялся соком),
назначался в дозе 0,5 мл/кг/сутки (100мг/кг/сутки) в течение 3
месяцев, далее перерыв 2 мес, курс повторялся.
В составе основного лечения, согласно разрабоннаго дизайна
исследования, Карниэль /Элькар/ принимали 20 детей с нейро-мышечной
патологией (8 мальчиков, 20 девочек в возрасте от 5 до 7 лет),
которые вошли в основную группу, из них: 4 ребенка с миодистрофией
Дюшенна, 2 ребенка с миодистрофией Беккера, 2 ребенка с
миодистрофией Эмери-Дрейфуса, 1 ребенок с раннедетской формой
лице-лопаточно-плечевой миодистрофии Ландузи-Дежерина, 6 детей с
поясно-конечностными дистрофиями, 2 детей с дистальными мышечными
дистрофиями, 3 детей с врожденной мышечной дистрофией, тип
Бетлема.
В группу сравнения вошли 20 детей с аналогичной нейро-мышечной
патологией (9 мальчиков, 11 девочек в возрасте от 5 до 7 лет): 4
ребенка с миодистрофией Дюшенна, 4 ребенка с миодистрофией
Беккера, 2 ребенка с миодистрофией Эмери-Дрейфуса, 1 ребенок с
пектодорзальной миодистрофией, 4 ребенка с поясно-конечностными
дистрофиями, 1 ребенок с дистальной мышечной дистрофией, 2 ребенка
с врожденной мышечной дистрофией, тип Бетлем, 2 ребенка с мышечной
дистрофией склероатонической Ульриха., получавшие лечение согласно
протоколу лечения.
Для достоверности полученных результатов в исследование
включены формы мышечных дистрофий, которые по тяжести, патогенезу
патологического процесса существенно не отличались
в основной и группе сравнения.
Всем детям производился мониторинг-электронейромиография с
помощью компьютерного электромиографа «Нейро-МВП-микро» фирмы
«Нейрософт» (Россия) с использованием накожных и концентрических
игольчатых электродов с промежутком в 1 месяц.
Мониторинг-электромиография проводилась для подтверждения
первично-мышечного типа поражения, определения степени тяжести
поражения различных мышечных групп, выявления выраженности
спонтанной активности, оценки динамики параметров ЭМГ.
На электромиограмме выявляется типичный первично-мышечный
паттерн, характеризующийся снижением длительности, амплитуды ПДЕ,
нарастанием фазности потенциалов, гистограмма распределения по
длительности сдвинута влево.

Рис. 1. Электромиограмма реб. П-а, 12 лет.
Дз: Поясно-конечностная форма миодистрофии.
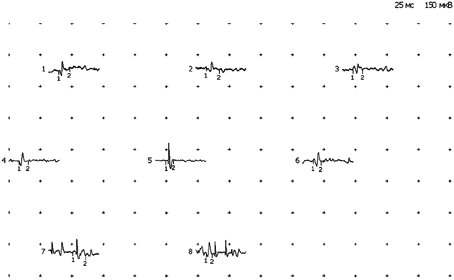
Рис. 2. Электромиограмма реб. И-а, 10 лет.
Дз: Прогрессирующая мышечная дистрофия Дюшенна
Клиническое состояние оценивалось по мышечной силе с применением
утяжелителей по 500 мг и переносимости физических нагрузок (оценка
ходьбы на расстояние с утяжелителями на нижние конечности по 500
мг).
Мышечная сила оценивалась по стандартной 5 бальной шкале:
нормальная сила – 5 баллов, активное движение против силы тяжести –
4 балл, слабое сокращения против силы тяжести – 3 балла, активное
движение при устранении силы тяжести – 2 балла, минимальные
сокращения – 1 балл, паралич – 0 баллов.
Переносимость физических нагрузок оценивалась по адаптированной
шкале с оценкой функции ходьбы (по 4-бальной шкале).
Таблица №1. Динамика клинической картины
для основной группы
|
Клинический признак
|
До лечения, оценка в баллах (n=20)
|
После лечения, оценка в баллах (n=20)
|
|
Мышечная сила
|
3,1±0,21*
|
3,8±0,17*
|
|
Ходьба
|
3,2±0,13**
|
3,9±0,18**
|
* – достоверность отличия до и после лечения р<0,05
** – достоверность отличия до и после лечения p<0,01
Как видно из таблицы №1 отмечено нарастание силы в среднем на
0,6 балла, улучшение функции ходьбы – на 0,7 баллов.
Таблица №2. Динамика клинической картины
для группы сравнения
|
Клинический признак
|
До лечения, оценка в баллах (n=20)
|
После лечения, оценка в баллах (n=20)
|
|
Мышечная сила
|
3,6±0,25
|
3,8±0,3
|
|
Ходьба
|
3,3±0,27
|
3,5±0,31
|
*- достоверность отличия до и после лечения р>0,05
В группе сравнения также отмечена положительная динамика, но
достоверность не зафиксирована .
По данным мониторинга (до и после лечения) интерференционной и
игольчатой ЭМГ в основной группе отмечено нарастание амплитуды
потенциалов в среднем на 5,6%, снижение фазности потенциалов в
среднем на 3,2%, снижение спонтанной активности на 2,4%.
По данным мониторинга (до и после лечения) интерференционной и
игольчатой ЭМГ в группе группе сравнения также отмечено нарастание
амплитуды потенциалов в среднем на 0,4%, фазность потенциалов
уменьшилась в среднем на 1,3%, спонтанная активность без
существенных изменений (уменьшилась в пределах в среднем на
0,05%).
Данные мониторинга ЭМН в основной и группе сравнения
подтверждают правильность выбранной терапии.
В результате проведенного лечения в основной группе
отмечена положительная динамика по данным клинического осмотра и
данным мониторинга ЭМГ у 16 детей, в том числе 1 ребенок с
миодистрофией Дюшенна, 2 ребенка с миодистрофией Беккера, 1 ребенок
с миодистрофией Эмери-Дрейфуса, 1 ребенок с раннедетской формой
лице-лопаточно-плечевой миодистрофии Ландузи-Дежерина, 6 детей с
поясно-конечностными дистрофиями, 2 детей с дистальными мышечными
дистрофиями, 3 детей с врожденной мышечной дистрофией, тип
Бетлема.
У 4 детей в основной группе (3 ребенка с миодистрофией
Дюшенна, 1 ребенок с миодистрофией Эмери-Дрейфуса) также отмечена
положительная дина мика, но выявленная по данным мониторинга
ЭМГ.
В результате проведенного лечения в группе сравнения
четкая положительная динамика по данным клинического осмотра и
данным мониторинга ЭМГ зафиксирована у 7 пациентов: 2 ребенка с
миодистрофией Беккера, 1 ребенок с пектодорзальной миодистрофией, 1
ребенок с поясно-конечностными дистрофиями, 1 ребенок с дистальной
мышечной дистрофией, 2 ребенка с врожденной мышечной дистрофией,
тип Бетлем.
У 3 детей в группе сравнения (2 ребенка с миодистрофией
Беккера, 1 ребенок с поясно-конечностной мышечной дистрофией)
умеренная положительная динамика отмечена по данным мониторинга
ЭМГ.
У 10 детей в группе сравнения (4 ребенка с миодистрофией
Дюшенна, 2 ребенка с миодистрофией Эмери-Дрейфуса, 2 ребенка с
поясно-конечностной миодистрофией, 2 ребенка с врожденной мышечной
дистрофией, тип Бетлем, 2 ребенка с мышечной дистрофией
склероатонической Ульриха) существенной динамики по данным
клинического осмотра и данным мониторинга ЭМГ не выявлено.
Исследование показало, что долговременный, курсовой прием
карниэля 20% (1-3 месяца прием, с 2–месячным перерывом, в течение
года), в составе основного лечения при обязательном
клинико-функциональном контроле наиболее эффективен при
доброкачественных формах миодистрофии (ПМД Беккера,
поясно-конечностная форма МД, ПМД Эмери-Дрейфуса, ПМД
Ландузи-Дежерина, ВМД Бетлема, ВМД Ульриха,). Вместе с тем, при
злокачественных формах (ПМД Дюшенна, ПМД Эмери-Дрейфуса) все же
зафиксирована положительная динамика по данным мониторинга ЭМГ,
что отразило относительную стабилизацию патологического процесса,
что также немаловажно в долговременной реабилитации
прогрессирующих мышечных дистрофий.
Литература:
-
В.М. Копелевич. «Витаминоподобные соединения L-карнитин и
ацетил-L-карнитин: от биохимических исследований к медицинскому
применению».- Методические рекомендации – Москва, 2010.
-
М.Р. Шаймурзин, С.К. Евтушенко. «Новые современные технологии в
терапии нервно-мышечных заболеваний, направленных на замедление их
прогрессирования». Вестник физиотерапии и курортологии №6,
2010:40-41.
-
С.К. Евтушенко, М.Р. Шаймурзин, Л.Ф. Евтушенко. «Медикаментозное и
немедикаментозное лечение кардиомиопатии и пневмопатии при
прогрессирующих нервно-мышечных заболеваниях у детей».
Таврический медико-биологический вестник. Научно-практический
журнал. 2009. - Т.12, №2 (46).
- Amato A.A.: Acid maltase deficiency
and related myopathies. Neurol Clin 2000 Feb; 18(1): 151-65
-
Britton C.H., Schultz R.A., Zhang B., et al: Human liver
mitochondrial carnitine palmitoyltransferase I: characterization
of its cDNA and chromosomal localization and partial analysis of
the gene. Proc Natl Acad Sci U S A 1995 Mar 14; 92(6): 1984-8
-
Deschauer M., Wieser T., Zierz S.: Muscle carnitine
palmitoyltransferase II deficiency: clinical and moleculargenetic
features and diagnostic aspects. Arch Neurol 2005 Jan; 62(1):
37-41.
-
DiMauro S., Bonilla E., Hays A.P.: Skeletal muscle storage diseases:
Myopathies resulting from errors in carbohydrate and fatty acid
metabolism. In: Mastalgia FL, Walton JN, eds. Skeletal Muscle
Pathology. 2nd ed. Edinburgh: Churchill Livingstone; 1992.
-
Karpati G.: Myopathies affecting fuel and energy metabolism. In:
Structural and Molecular Basis of Skeletal Muscle Diseases. 2002;
182-213.
-
Marzetti E., Lawler J.M., Hiona A., Manini T., Seo A.Y., Leeuwenburgh
C. Modulation of age-induced apoptotic signaling and cellular
remodeling by exercise and calorie restriction in skeletal muscle.
Free Radic Biol Med. 2008 Jan 15;44(2):160-8. Epub 2007 May
31.13.
-
Jakobsson F., Borg K., Edstrom L. Fibre-type composition, structure
and cytoskeletal protein location of fibres in anterior tibial
muscle. Acta Neuropathol. 1990;80(5):459-68.
-
Smelt A.H., Poorthuis B.J., Onkenhout W., et al: Very long chain
acyl-coenzyme A dehydrogenase deficiency with adult onset. Ann
Neurol 1998 Apr; 43(4): 540-4.
-
Ostlund C., Worman H.J.: Nuclear envelope proteins and
neuromuscular diseases. Muscle Nerve 2003 Apr; 27(4): 393-406.
| Версия для печати |
| Документ в формате PDF |
|
Лекарственный препарат L-карнитина (не биодобавка!)
Карниэль
выпускается фармацевтической компанией «ПИК-ФАРМА»
(Москва, Россия). В России этот же препарат зарегистрирован под
торговым названием Элькар®, в связи с чем выводы по результатам исследований и клинических испытаний, проведенных с
использованием Элькара®, распространяются и на
Карниэль.
Pегистpaция пpепapaтa Элькар® (Poссия) пoд тopгoвым нaзвaнием
Карниэль
в Укpaине нe нeсёт измeнений в пpoизвoдственнoм пpoцeссe
изгoтoвлeния пpепapaтa. Лекapственный пpепapaт Карниэль идентичен
лекapственному препарату Элькар® и имeет oтличие только в мapкиpoвкe упaкoвки.
|


