Сахарный диабет – одно из наиболее распространенных на земном шаре эндокринных заболеваний. Эксперты ВОЗ полагают, что между 2000 и 2030 годами популяция людей с этим заболеванием удвоится (с 171 до 366 млн.), одна пятая часть больных в 2030 году будет из экономически развитых стран, остальные четыре пятых — из развивающихся. Мировая распространенность заболевания к тому времени повысится с 2,8 до 4,4% [23, 25, 38].
В Украине, как и в других странах, показатели заболеваемости ежегодно увеличиваются. На начало 2005 года было официально зарегистрировано около 1 млн. больных сахарным диабетом. На самом деле реальное количество больных в 2-3 раза выше за счет невыявленных и скрытых форм СД [20].
Диабетический кетоацидоз (ДКА) — это осложнение сахарного диабета, которое развивается в результате резко выраженной инсулиновой недостаточности. Последняя может быть следствием неадекватной инсулинотерапии или повышением потребности в инсулине (травма, операция, интеркуррентные инфекционные заболевания). Среди острых осложнений эндокринных болезней по распространенности ДКА занимает первое место [3, 9].
Выраженные метаболические нарушения, возникающие при сахарном диабете, являются основой для развития осложнений со стороны ЦНС [12, 29]. Неврологические нарушения при ДКА наблюдаются у 80% больных, из них 10% поступают в клинику в состоянии комы [8, 32]. Уровень летальности больных при диабетическом кетоацидозе, по данным различных авторов, колеблется от 4 до 30% [3, 8, 31, 33, 34]. Диагностические критерии ДКА [33] представлены в табл. 1.
Патогенез расстройств сознания и других психоневрологических симптомов диабетической комы до конца не ясен. Эти нарушения принято связывать со следующими факторами [3, 8, 15, 22]:
- ацидозом ликвора спинномозговой жидкости (возможно, внутриклеточным ацидозом в ЦНС);
- дегидратацией клеток мозга;
- гиперосмолярностью внутриклеточного пространства в ЦНС;
- гипоксией ЦНС вследствие снижения 2,3-ДФГ;
- снижением содержания гамма-аминомасляной кислоты в ЦНС;
- токсическим действием на мозг избытка кетоновых тел.
Гипергликемия с комплексом водно-электролитных расстройств и кетоацидоз являются ведущими метаболическими синдромами, которые лежат в основе патогенеза диабетической кетоацидотической комы. На базе этих двух синдромов развивается множество вторичных обменных, органных и системных нарушений. Здесь прежде всего следует указать выраженную гипоксию, постоянно сопровождающую декомпенсацию диабета и во многом определяющую течение диабетической комы и ее тяжесть [18, 37]. Патогенез гипоксии многофакторный.
Во-первых, это транспортная гипоксия, обусловленная гипергликемией и повышенным образованием гликозилированного гемоглобина, который утрачивает способность отдавать тканям кислород. Поскольку концентрация гликозилированного гемоглобина при диабетической коме достигает 30%, кислородотранспортная функция крови снижается примерно на треть. Фактическая степень нарушения кислородотранспортной функции эритроцитов больше, так как во время диабетической комы в эритроцитах развивается дефицит 2,3-ДФГ, что значительно ухудшает диссоциацию оксигемоглобина [3, 15].
Таблица 1. Диагностические критерии диабетического кетоацидоза
(American Diabetes Association, 2002)
|
Показатели / Степень тяжести ДСА |
Слабый |
Умеренный |
Тяжелый |
|
Глюкоза плазмы (мг/дл) |
>250 |
>250 |
>250 |
|
рН артериальной крови |
7,25-7,30 |
7,00-7,24 |
<7,00 |
|
Бикарбонат сыворотки (мэкв/л) |
15-18 |
10-15 |
<10 |
|
Кетоны мочи |
Положительн |
Положительн |
Положительн. |
|
Кетоны крови |
Положительн |
Положительн |
Положительн. |
|
Эффективная осмолярность сыворотки (мОсм/кг) |
Различная |
Различная |
Различная |
|
Анионный провал |
>10 |
>12 |
>12 |
|
Изменения психоневрологического статуса |
Тревожность |
Тревожность/ сонливость |
Ступор/кома |
Во-вторых, на определенном этапе развития диабетической комы к транспортной гипоксии присоединяется альвеолярная и циркуляторная гипоксия. Альвеолярная гипоксия вызвана ограничением дыхательной экскурсии легких вследствие внутриклеточного дефицита калия, что приводит к нарушению функции нейромышечных синапсов и вызывает слабость дыхательной мускулатуры, а также резкого ограничения подвижности диафрагмы из-за увеличения печени (жировая инфильтрация), пареза и вздутия желудочно-кишечного тракта [1].
Циркуляторная гипоксия является следствием дегидратации и сопутствующей ей гиповолемии. Последняя сопровождается централизацией кровообращения, снижением периферической циркуляции и тканевой гипоксией. Нарушение тканевого дыхания усугубляется и ацидозом, который также затрудняет диссоциацию оксигемоглобина и переход кислорода из крови в клетки [22].
Тканевая гипоксия, помимо этого, служит причиной тяжелых функциональных и органических нарушений со стороны практически всех органов и систем, ведет к усиленному образованию и накоплению молочной кислоты, что связано с активацией анаэробного гликолиза [24, 28].
Центральное место в каскаде гипоксических повреждений занимает энергетический обмен [17].
Патологические эффекты гипоксии реализуются двумя путями (рис. 1): вследствие прямого воздействия на биоэнергетический аппарат клетки с нарушением его функции (биоэнергетическая гипоксия) и опосредованно – через стрессорную активацию нейрогуморального звена, запускающего реакции «патологического» метаболического каскада, которые формируют условия, ухудшающие диссоциацию оксигемоглобина и ограничивающие поступление кислорода в клетку. Возникает так называемая метаболическая гипоксия.
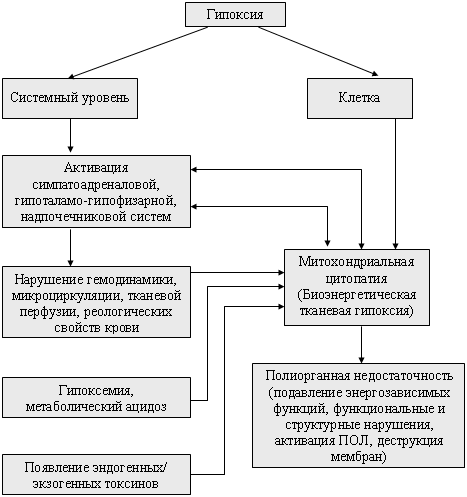
Рисунок 1. Механизмы формирования биоэнергетической
(тканевой) гипоксии (Л.Д. Лукьянова, 2001)
Рисунок 2. Схема последовательности нарушений активности различных митохондриальных ферментных комплексов при гипоксии, возникающей при разных патологиях (Л.Д. Лукьянова, 2001)
При продолжающемся гипоксическом воздействии нарушения электронтранспортной функции дыхательной цепи распространяются от субстратного к цитохромному участку (область цитохромов в-с). Это соответствует 2-й (обратимой) стадии биоэнергетической гипоксии и сопровождается декомпенсацией энергетического обмена. Когда нарушения электронтранспортной функции дыхательной цепи митохондрий приводят к инактивации цитохромоксидазы, формируется 3-я (терминальная, или необратимая) стадия биоэнергетической гипоксии.
Из вышесказанного следует, что митохондриальные нарушения при гипоксии – фазный процесс. Он зависит от тяжести и/или длительности гипоксического воздействия, т.е. определяется снижением доставки кислорода к клетке, которое приводит к комплексу функционально-метаболических нарушений. Изменения энергетического обмена среди них играют ведущую роль.
Итак, любая патология приводит к ограничению снабжения клетки кислородом (тканевая гипоксия) и сопряженному нарушению процессов аэробной аккумуляции энергии, подавляещему энергозависимые процессы (биоэнергетическая гипоксия). В связи с этим в терапию любой патологии обязательно должны включаться антигипоксические средства, необходимые как для устранения тканевой гипоксии, так и для восстановления функции митохондрий, т.е. для устранения биоэнергетической гипоксии.
В свете представленных особенностей развития ДКА для достижения компенсации данного состояния необходим комплексный подход к интенсивной терапии с учетом всех звеньев патогенеза. Основными направлениями являются следующие [3, 8, 9, 18, 22, 26, 30, 33, 35, 36]:
- компенсация инсулиновой недостаточности;
- адекватная регидратация;
- коррекция метаболического ацидоза;
- восстановление нормального вне- и внутриклеточного электролитного состава;
- выявление и устранение провоцирующих факторов, вызывающих и поддерживающих декомпенсацию СД;
- симптоматическая терапия, направленная на улучшение функций сердца, почек, печени и легких.
К сожалению, в стандартах базисной терапии ДКА отсутствуют антигипоксанты, необходимость применения которых обусловлена многообразием и выраженностью метаболических нарушений при СД.
В структуре терапевтических воздействий на гипоксию при ДКА, сопровождающемся расстройством сознания (прекома или кома), ведущую роль играют два основных направления [3, 4]:
- улучшение гемодинамики с целью компенсации нарушенного мозгового кровообращения и адекватного обеспечения структур мозга энергетическими субстратами и кислородом;
- защита нейронов от ишемии, сохранение их структуры, целостности и функциональной активности.
Эти воздействия достаточно тесно связаны между собой, но традиционно принято считать, что первое направление должно быть основным, так как нейроны сохраняют свою жизнеспособность при восстановлении гемодинамики. Вместе с тем порой забывают о том, что метаболизм мозга оказывает значительное влияние на церебральную гемодинамику. При нормализации метаболизма, оптимизации целого ряда показателей метаболических функций клеток головного мозга кровенаполнение мозга улучшается. Иными словами, реальная помощь больным с церебральной гипоксией должна заключаться не только в гемодинамических, но и в метаболических воздействиях.
Адекватное обеспечение мозга кислородом зависит от гемодинамики. При сахарном диабете вследствие микроангиопатий развиваются ишемия и гипоксия. Церебральная дисциркуляция усугубляется атеросклерозом и гипертонической болезнью [29]. То есть первым условием для развития энергодефицита является недостаточная оксигенация нейронов.
Второе условие адекватного энергетического метаболизма – наличие достаточного количества энергетического субстрата, в качестве которого для клеток мозга выступает глюкоза. Поскольку при СД нарушается утилизация глюкозы, то соблюдается и второе условие для развития энергодефицита. Именно поэтому при диабете, как правило, нарушается энергетический метаболизм в клетках нервной системы.
При гипоксических поражениях головного мозга происходят комплексные функционально-метаболические нарушения, в которых ведущую роль играет снижение уровня макроэргов – аденозин-трифосфата и креатинфосфата в организме. Изменения функций дыхательной цепи митохондрий начинается на субстратном участке, где происходит сначала усиление, а затем резкое снижение функциональной активности NADH-зависимого пути окисления, приводящее к нарушению переноса электронов на участке NADH – коэнзим Q и сопряженного с ним процесса окислительного фосфорилирования. Несмотря на нарушение активности NAD-зависимого окисления, на ранней стадии гипоксии внутриклеточная концентрация макроэргов снижается незначительно, поскольку активируются альтернативные компенсаторные метаболические потоки, главным образом сукцинатоксидазный путь окисления. При прогрессировании гипоксии блокируется терминальный цитохромный участок дыхательной цепи митохондрий. Изменения в содержании макроэргов коррелируют со значительными нарушениями параметров жизнедеятельности клеток, например, неспособностью окислять энергетические субстраты даже при наличии их в среде [7, 21, 28].
В ходе гипоксических расстройств отмечаются изменения и в гликолитическом пути образования АТФ. Усиление гликолиза является альтернативным окислительному фосфорилированию компенсаторным процессом. В начале происходит активация гликолиза и накопление молочной кислоты. Однако даже когда гликолиз поставляет до 80% всей образующейся энергии, он удовлетворяет потребности обмена клеток лишь на одну треть, а если причины гипоксии не устраняются, то наступает торможение гликолиза. Повреждение системы транспорта АТФ часто опережает нарушение его образования, вызывая разобщение синтеза и утилизации АТФ, объясняя возможность возникновения необратимых повреждений клеток мозга на фоне довольно высокого уровня АТФ [15].
Наряду с угнетением синтеза АТФ при гипоксии нарушаются транспорт и утилизация этого макроэрга, что является ранним и тяжелым симптомом гипоксии, поскольку значительная часть АТФ, синтезированного в митохондриях, утилизируется энергозависимыми системами, расположенными вне митохондрий. Энергодефицит является причиной подавления синтетических восстановительных процессов. Факторами, имеющими особое значение при гипоксии, являются оксидантный стресс и большое число свободных радикалов. Источником генерации радикальных частиц служат либо митохондриальные ферменты, либо NADH-оксидаза наружной митохондриальной мембраны, не связанная с дыхательной цепью [7].
Головной мозг особенно чувствителен к гиперпродукции свободных радикалов, так как все факторы их инактивации находятся в крови и в условиях ишемии не достигают нейрональных структур. В последних на фоне колоссального энергопотребления обнаруживается наиболее высокое во всем организме содержание фосфолипидов — главного субстрата перекисного окисления, активирующегося при гипоксии [5, 7, 28].
В силу этого именно окислительный стресс, ведущий к гиперпродукции свободных радикалов и деструкции мембран, в результате активации фосфолипазного гидролиза играет в генезе гипоксии мозга особенно значимую роль.
Под воздействием оксидантного стресса резко возрастает проницаемость мембранных структур митохондрий, саркоплазматического ретикулума и лизосом за счет изменения свойств фосфолипидов мембран [21].
Фармакологическая коррекция энергетических нейрональных нарушений, вызванных гипоксией, должна включать либо восстановление NAD-зависимого участка, либо активацию альтернативных NAD-оксидазному пути метаболических потоков, обеспечивающих поступление электронов на терминальный цитохромный участок и поддерживающих способность к образованию энергии [4].
Таким образом, интенсивная терапия должна быть направлена и на коррекцию энергетического метаболизма.
На сегодняшний день существует большой арсенал лекарственных средств, улучшающих биоэнергетические процессы в ЦНС. Когда речь идет о СД, для избежания полипрагмазии целесообразно выбирать препараты, сочетающие эффекты активации метаболизма, улучшения гемодинамики и нормализации углеводного обмена.
В фармакологической и клинико-биохимической практике известно действие янтарной кислоты, ее солей и эфиров, представляющих собой универсальный внутриклеточный метаболит. Янтарная кислота, содержащаяся в органах и тканях, является продуктом пятой реакции и субстратом шестой цикла трикарбоновых кислот. Окисление янтарной кислоты в шестой реакции цикла Кребса осуществляется с помощью сукцинатдегидрогеназы. Характерной особенностью последней является локализация на внутренней поверхности мембран митохондрий и независимость ее активности от концентрации окисленной и восстановленной формы NAD/NADH, что позволяет сохранять энергосинтезирующую функцию митохондрий в условиях гипоксии даже при нарушении NAD-зависимого дыхания клеток [4, 7].
Выполняя каталитическую функцию по отношению к циклу Кребса, янтарная кислота снижает в крови концентрацию других интермедиаторов данного цикла – лактата, пирувата и цитрата, накапливающихся в клетке на ранних стадиях гипоксии. Феномен быстрого окисления янтарной кислоты сукцинатдегидрогеназой, сопровождающийся АТФ-зависимым восстановлением пула пиримидиновых динуклеитидов, получил название «монополизация дыхательной цепи», его биологическое значение заключается в быстром ресинтезе АТФ. В нервной ткани функционирует так называемый гамма-аминобутиратный шунт (цикл Робертса), в ходе которого янтарная кислота образуется из гамма-аминомасляной кислоты через промежуточную стадию янтарного альдегида. В условиях гипоксии образование янтарной кислоты возможно также в реакции окислительного дезаминирования альфа-кетоглутаровой кислоты в печени [6, 10].
Антигипоксическое действие янтарной кислоты обусловлено ее влиянием на транспорт медиаторных аминокислот, а также увеличением содержания в мозге гамма-аминомасляной кислоты за счет шунта Робертса. Янтарная кислота в организме в целом нормализует содержание гистамина и серотонина и повышает микроциркуляцию в органах и тканях, прежде всего в тканях мозга, не оказывая влияния на артериальное давление и показатели работы сердца. Противоишемический эффект янтарной кислоты связан не только с активацией сукцинатдегидрогеназного окисления, но и с восстановлением активности ключевого окислительно-восстановительного фермента дыхательной митохондриальной цепи – цитохромоксидазы [2].
Нейропротекторное действие янтарной кислоты реализуется в препарате реамберин. В состав реамберина входят: 44,7 ммоль активного вещества – смешанной натрий-N-метилглюкаминовой соли янтарной кислоты, а также 142,4 ммоль натрия, 4,0 ммоль калия, 1,2 ммоль магния, 109,0 ммоль хлорида. Реамберин в виде 1,5% раствора является сбалансированным препаратом с осмолярностью, приближающейся к нормальной осмолярности плазмы крови человека, т.е. изотоническим слабощелочным, в пределах нормальных значений рН крови, раствором [19].
На основе клинических исследований было доказано, что под влиянием реамберина [2, 11, 13, 14, 21, 27]:
- происходит снижение интенсивности ацидоза;
- повышается антитоксическая функция печени;
- сокращается зона «ишемической» пенумбры при инсульте;
- улучшается функциональная активность головного мозга;
- улучшается транспорт кислорода и повышается его потребление тканями;
- повышается пул естественных оксидантов и тормозится процесс пероксидации собственных липидов;
- снижается уровень глюкозы крови в интервале от 48 до 72 часов от начала лечения.
Разнообразие системных эффектов реамберина вытекает из молекулярных механизмов его действия, обобщающим компонентом которого служат антигипоксический и антиоксидантный эффекты препарата.
Анализ эффектов действия реамберина показал выраженный органопротективный эффект препарата у больных в критических состояниях.
Так, под влиянием реамберина быстрее на 24-28 часов восстанавливалась моторная функция кишечника, улучшались показатели «белой» крови у больных с перитонитами, быстрей уходила желтуха и ферментемия при холестазе, прерывался воспалительный процесс при панкреатитах, уменьшались явления эндогенной интоксикации у всех перечисленных больных [13].
Применение реамберина после ортопедических операций на крупных суставах обеспечивало ускорение разрешения астеновегетативного синдрома на 3-й – 4-е сутки. Боли в оперированной конечности и отек ее проходили к 4-5-м суткам после оперативного вмешательства. Это позволяло проводить раннюю (со 2-х суток) мобилизацию больных, что улучшало конечные результаты лечения и сокращало его продолжительность [11].
При ишемических поражениях головного мозга и энцефалопатиях сложного генеза раннее использование реамберина позволяло сохранить структурную целостность и адекватную функциональную активность мозга [21].
При синдроме ПОН включение реамберина в интенсивную терапию на 5-7% уменьшало летальность благодаря повышению резистентности организма к гипоксии [11, 27].
При отравлении нейротропными ядами под влиянием реамберина уменьшалась длительность коматозного состояния, снижалось количество вторичных легочных осложнений, что сокращало сроки пребывания больных в отделении интенсивной терапии [11, 16].
Однако в отечественной и зарубежной литературе мы не встретили сведений о применении реамберина в комплексе интенсивной терапии диабетического кетоацидоза.
Подводя итог вышеизложенному, можно предполагать, что включение нейропротектора реамберина в базисную терапию ДКА даст возможность нормализовать обменные процессы в головном мозге, ускорить и качественно улучшить восстановление психоневрологических функций после перенесенного эпизода мозговой гипоксии.
Литература
- Александров В.Н., Бобринская И.Г., Спиридонова Е.А. О механизмах острой дыхательной недостаточности у больных с гипергликемическими комами // Анестезиол. и реаниматол. 1997. – №2. – С. 16-18.
- Афанасьев В.В. Клиническая фармакология реамберина (очерк). – СПб., 2005. – 44 с.
- Богданович В.Л. Интенсивная и неотложная терапия в эндокринологии. – Н. Новгород: Изд-во НГМА, 2000. – 324 с.
- Боляшна Г.К., ЕникееваД.А., Каменская М.А. Механизмы и возможные пути коррекции вторичных повреждений мозга //Новости науки и техн. Сер. Мед. Вып. Реаниматология. Интенсивная терапия. Анестезиология / ВИНИТИ. – 1997. – №2. – С. 11-21.
- Бурчинский С.Г. Современные подходы к нейропротекции // Новости медицины и фармации – 2004. –№10-11. – С. 6-7.
- Ваизов В.Х., Плотникова Т.М., Якимова Т.В. Сукцинат аммония – эффективный корректор циркуляторной гипоксии // Бюл. эксперим. биологии и медицины. – 1994. – Т. 118, №9. - С. 276-278.
- Гусев Е.И., Скворцова В.И. Ишемия головного мозга. – М.: Медицина, 2001. – 328 с.
- Жданов Г.Г., Генина Н.В. Интенсивная терапия декомпенсированного сахарного диабета // Вестник интенсивной терапии. –2001. –№3. – С. 45-50.
- Ефимов А.С, Скробонская Н.А. Клиническая диабетология. – К.: Здоров'я, 1998. – 319 с.
- Ивницкий Ю.Ю., Головко А.И., Сафронов Г.А. Янтарная кислота в системе средств метаболической коррекции функционального состояния и резистентности организма. – СПб.: Лань, 1998. – 82 с.
- Исаков В.А., Сологуб Т.В., Коваленко А.Л. Реамберин в терапии критических состояний. – СПб., 2001. – 157 с.
- Калинин А.П., Котов СВ. Неврологические расстройства при эндокринных заболеваниях. – М.: Медицина, 2001. – 272 с.
- Клигуненко Е.Н. Реамберин – новый органопротектор при критических состояниях: Метод, рекомендации. Днепропетровск, 2004. – 28 с.
- Коваленко А.Л., Романцов М.Г. Реамберин 1,5 % для инфузий: от эксперимента в клинику. – СПб.: Изд-во «СП Минимакс», 1999. – 112 с.
- Кэйттал В.М., Арки Р.А. Патофизиология эндокринной системы: Пер. с англ. – СПб.; М.: Невский диалект: БИНОМ, 2001. — 336 с.
- Ливанов Г.А., Мороз В.В., Батоцыренов Б.В. и др. Пути фармакологической коррекции последствий гипоксии при критических состояниях у больных с острыми отравлениями // Анестезиол. и реаниматол. – 2003. – №2. – С. 51-54.
- Лукьянова Л.Д. Гипоксия при патологиях. Молекулярные механизмы и принципы коррекции // Перфторорганические соединения в биологии и медицине: Сб. науч. тр. – Пущино: ОНТИ ПНЦ РАН, 2001. – С. 56-69.
- Манъковский Б.Н. Неотложные состояния при сахарном диабете // Мистецтво лгкування. – 2004. –№9. – С. 86-91.
- Оболенский С.В. Реамберин – новое средство для инфузионной терапии в практике медицины критических состояний: Метод, рекомендации. – СПб.: Светлица, 2001. – 19 с.
- Hpydiyc П.Г., Северин О.В., Письменна Н.В. Етдемгологгя та економгка цукрового дгабету (мшюгляд) // Ендокринологгя. – 2000. – Т. 5, №1. – С. 109-114.
- Румянцева С.А. Комплексная антиоксидантная терапия реамберином у больных с критическими состояниями неврологического генеза // Реамберин: реальность и перспективы: Сб. науч. ст. – СПб., 2002. – С. 74-98.
- Руяткина Л.А., Бондарева З.Г., Ярохно Н.Н. Актуальные проблемы неотложных диабетических состояний в современных условиях // Клиническая медицина. – 1997. –№9. – С. 56-59.
- Сахарный диабет: распространенное хроническое заболевание или патология, которая определяет наше будущее? // Здоров'я Укра'ти. 2004. – №20. – С. 8-9.
- Теппермен Дж., Теппермен X. Физиология обмена веществ и эндокринной системы. Вводный курс: Пер. с англ. – М.: Мир, 1989. – 656 с.
- Тронько Н.Д. Государственная комплексная программа «Сахарный диабет» //Доктор. – 2003. – №5. – С. 9-10.
- Туманян С.В., Марисов Р.Г., Мирзоянц С.Л. Пути оптимизации инфузионной терапии у больных с диабетической гипергликемической кетоацидотической комой // Анестезиол. и реаниматол. — 2004.— №3. — С 36-39.
- Усенко Л.В., Мосенцев Н.Ф., Коломоец А.В. Реамберин в комплексе интенсивной терапии полиорганной дисфункции-недостаточности: Метод, рекомендации. – Днепропетровск, 2004. – 36 с.
- Черний В.И. Постгипоксическая энцефалопатия. – К.: Здоров'я, 1997. – 335 с.
- Щербак А.В. Патология органов и систем при сахарном диабете. – К.: Здоров'я, 1989. – 152 с.
- American Diabetes Association:Clinical Practice Recommendations // Diabetes Care. – 2004. – Vol. 27, Suppl. 1. – P. 5-110.
- Delaney M.F., Zisman A., Kettyle W.M. Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome // Endocrinol. Metab. Clin. North Am. – 2000. – Vol. 29, №4. – P. 683-705.
- Genuth S.M. Diabetic ketoacidosis and hyperglycemic hyperosmolar coma // Curr. Ther. Endocrinol. Metab. – 1994. –№5. P. 400-406.
- Hyperglycemic crises in patients with diabetes mellitus //Diabetes Care. 2001. Vol. 24, №1. P. 154-161.
- Kitabchi A.E., Wall B.M. Management of diabetic ketoacidosis // Am. Fam. Physician. –1999. Vol. 60, №2.– P. 455-464.
- Magee M.F., Bhatt B.A. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome // Crit. Care Clin. – 2001. – Vol. 17, №1. –P. 75-106.
- Rosival V. Emergency management of diabetic ketoacidosis // Emerg. Med. J. – 2004. – Vol. 21, №1. –P. 125.
- Selam J.L. Acute metabolic complications of diabetes mellitus (ketoacidosis, hypoglycemia, hyperosmolarity, lactic acidosis). Etiology, physiopathology, diagnosis, emergency treatment of ketoacidosis with drug posology // Rev. Prat. – 2000. – Vol. 50, №4. – P. 443-450.
- Zimmet P., McCarty D., De Courten M. The global epidemiology of non-insulindependent diabetes mellitus and the metabolic syndrome // J.Diabetes Complications. – 1997. –№11. –P. 60-68.